One of the most important weak interactions between biologically important molecules is the hydrogen bond (H-bond). These “bonds” are the result of electrostatic attraction caused by the uneven distribution of electrons within covalent bonds. For example, the bonding electron pairs of the H–O bonds of water molecules are attracted more tightly to the oxygen atoms than to the hydrogen atoms. A small net positive charge is left on the hydrogen and a small net negative charge on the oxygen. Such polarization of the water molecules can be indicated in the following way:

Here the δ+ and δ– indicate a fraction of a full charge present on the hydrogen atoms and on the nonbonded electron pairs of the oxygen atom, respectively. Molecules such as H2O, with strongly polarized bonds, are referred to as polar molecules and functional groups with such bonds as polar groups. They are to be contrasted with such nonpolar groups as the –CH3 group in which the electrons in the bonds are nearly equally shared by carbon and hydrogen.
A hydrogen bond is formed when the positively charged end of one of the dipoles (polarized bonds) is attracted to the negative end of another dipole. Water molecules tend to hydrogen bond strongly one to another; each oxygen atom can be hydrogen-bonded to two other molecules and each hydrogen to another water molecule. Thus, every water molecule can have up to four hydrogen-bonded neighbors.
 A water molecule hydrogen bonded to four other water molecules; note the tetrahedral arrangement of bonds around the central oxygen.
A water molecule hydrogen bonded to four other water molecules; note the tetrahedral arrangement of bonds around the central oxygen.
Many groups in proteins, carbohydrates, and nucleic acids form hydrogen bonds to one another and to surrounding water molecules. For example, an imidazole group of a protein can bond to an OH group of an amino acid side chain or of water in the following
ways:

Remember that hydrogen bonds are always formed between pairs of groups, with one of them, often C=O or C=N-, containing the negative end of a dipole and the other providing the proton. The proton acceptor group, often OH or NH and occasionally SH, and even CH in certain structures,donates an unshared pair of electrons. Dashed arrows are sometimes drawn from the hydrogen atom to the electron donor atom to indicate the direction of a hydrogen bond. Do not confuse these arrows with the curved arrows that indicate flow of electrons in organic reactions.
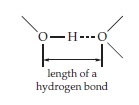
The strength of hydrogen bonds, as measured by the bond energy, varies over the range 10–40 kJ/mol. The stronger the hydrogen bond the shorter its length. Because hydrogen atoms can usually not be seen in X-ray structures of macromolecules, the lengths of
hydrogen bonds are often measured between the surrounding heavy atoms:

A typical —OH- - -O hydrogen bond will have a length of about 0.31 nm; a very strong hydrogen bond may be less than 0.28 nm in length, while weak hydrogen bonds will approach 0.36 nm, which is the sum of the van der Waals contact distances plus the O–H bond length. Beyond this distance a hydrogen bond cannot be distinguished easily from a van der Waals contact.
Hydrogen bonds are strongest when the hydrogen atom and the two heavy atoms to which it is bonded are in a straight line. For this reason hydrogen bonds tend to be linear. However, the dipoles forming the hydrogen bond do not have to be colinear for strong hydrogen bonding: There is some preference for hydrogen bonding to occur in the direction of an unshared electron pair on the oxygen or nitrogen atom.

A linear O–H- - -O hydrogen bond with dipoles at an angle one to another.
Both ammonia, NH3, and the –NH2 groups of proteins are good electron donors for hydrogen bond formation. However, the hydrogen atoms of uncharged –NH2 groups tend to be poor proton donors for H-bonds. Do hydrogen bonds have some covalent character? The answer is controversial.
Hydrogen bonding is important both to the internal structure of biological macromolecules and in interactions between molecules. Hydrogen bonding often provides the specificity necessary to bring surfaces together in a complementary way. Thus, the
location of hydrogen-bond forming groups in surfaces between molecules is important in ensuring an exact alignment of the surfaces.37 The hydrogen bonds do not always have to be strong. For example, Fersht and coworkers, who compared a variety of mutants of an enzyme of known three-dimensional structure, found that deletion of a side chain that formed a good hydrogen bond to the substrate weakened the binding energy by only 2–6 kJ/mol. However, loss of a hydrogen bond to a charged group in the substrate caused a loss of 15–20 kJ/mol of binding energy. Study of mutant proteins created by genetic engineering is now an important tool for experimentally investigating the biological roles of hydrogen bonding.

 A “ribbon” drawing of the 307- residue proteinhydrolyzing enzyme carboxypeptidase A. In this type of drawing wide ribbons are used to show β strands and helical turns while narrower ribbons are used for bends and loops of the peptide chains. The direction from the N terminus to C terminus is indicated by the arrowheads on the β strands. No individual atoms are shown and side chains are omitted. Courtesy of Jane Richardson.
A “ribbon” drawing of the 307- residue proteinhydrolyzing enzyme carboxypeptidase A. In this type of drawing wide ribbons are used to show β strands and helical turns while narrower ribbons are used for bends and loops of the peptide chains. The direction from the N terminus to C terminus is indicated by the arrowheads on the β strands. No individual atoms are shown and side chains are omitted. Courtesy of Jane Richardson.